
APRT Deficiency / 2,8-DHA crystalluria
Disease Information

Expand All Topics | Close All Topics
Adenine phosphoribosyltransferase (APRT) deficiency is an underrecognized, autosomal recessive disorder of adenine metabolism, leading to 2,8-dihydroxyadeninuria that causes nephrolithiasis and kidney failure in a significant proportion of untreated patients. The disorder has been found in all ethnic groups and 24 functionally significant APRT mutations have been reported to date [1]. Of more than 300 cases of APRT deficiency reported world-wide, more than 200 came from Japan, a substantial number of patients from France and Iceland [1], and about 15 cases have been reported in the US (personal communication A. Sahota). The estimated prevalence of APRT deficiency is 0.5 to 1 per 100,000 in the Caucasian population, 0.25 to 0.5 per 100,000 in the Japanese population and in Iceland the estimated point prevalence is 8.9/100,000. Possible explanations for the apparently low prevalence in countries other than Iceland and Japan, include lack of awareness of this disorder, inadequate evaluation of patients with kidney stones, and erroneous diagnosis of 2,8-dihydroxyadenine (2,8-DHA) stones as uric acid or xanthine stones as all three of these types of stones are radiolucent.
Although the majority of patients with APRT deficiency present with kidney stone disease there is tremendous variability in the clinical expression of APRT deficiency, ranging from an asymptomatic condition to life-threatening kidney failure [2-4]. In patients who have developed end-stage kidney failure the diagnosis of APRT deficiency has generally not been made prior to commencement of renal replacement thereapy [5, 6] and there are several reports in the literature of recurrent 2,8-DHA-induced nephropathy following kidney transplantation [4-10]. In our experience and that of other investigators, occasional patients complain of eye discomfort but it is not clear if these symptoms are related to 2,8-DHA crystal deposition in ocular tissues such as the cornea (personal communication A. Sahota, D. Milliner).
References
- Edvardsson Vidar PR. Adenine Phosphosribosyltransferase deficiency and 2,8-Dihydroxyadeninuria. In: Moriwaki. Y, ed. Genetic Errors Associated with Purine and Pyrimidine Metabolism in Humans: Diagnosis and Treatment Research Signpost; 2006.
- Simmonds HA. 2,8-Dihydroxyadenine lithiasis--epidemiology, pathogenesis and therapy. Verh Dtsch Ges Inn Med 1986;92:503-8.
- Edvardsson V, Palsson R, Olafsson I, Hjaltadottir G, Laxdal T. Clinical features and genotype of adenine phosphoribosyltransferase deficiency in iceland. Am J Kidney Dis 2001;38(3):473-80.
- Cassidy MJ, McCulloch T, Fairbanks LD, Simmonds HA. Diagnosis of adenine phosphoribosyltransferase deficiency as the underlying cause of renal failure in a renal transplant recipient. Nephrol Dial Transplant 2004;19(3):736-8.
- Brown HA. Recurrence of 2,8-dihydroxyadenine tubulointerstitial lesions in a kidney transplant recipient with a primary presentation of chronic renal failure. Nephrol Dial Transplant 1998;13(4):998-1000.
- Eller P, Rosenkranz AR, Mark W, Theurl I, Laufer J, Lhotta K. Four consecutive renal transplantations in a patient with adenine phosphoribosyltransferase deficiency. Clin Nephrol 2004;61(3):217-21.
- Benedetto B, Madden R, Kurbanov A, Braden G, Freeman J, Lipkowitz GS. Adenine phosphoribosyltransferase deficiency and renal allograft dysfunction. Am J Kidney Dis 2001;37(5):E37.
- de Jong DJ, Assmann KJ, De Abreu RA, et al. 2,8-Dihydroxyadenine stone formation in a renal transplant recipient due to adenine phosphoribosyltransferase deficiency. J Urol 1996;156(5):1754-5.
- Gagne ER, Deland E, Daudon M, Noel LH, Nawar T. Chronic renal failure secondary to 2,8-dihydroxyadenine deposition: the first report of recurrence in a kidney transplant. Am J Kidney Dis 1994;24(1):104-7.
- Glicklich D, Gruber HE, Matas AJ, et al. 2,8-dihydroxyadenine urolithiasis: report of a case first diagnosed after renal transplant. Q J Med 1988;68(258):785-93.
APRT deficiency should be considered in any patient presenting with renal colic, radiolucent urolithiasis and tubulointerstitial nephropathy of unknown cause, as well as in infants with a history of reddish-brown diaper stain [1, 2].
References
- Simmonds HA. 2,8-Dihydroxyadenine lithiasis--epidemiology, pathogenesis and therapy. Verh Dtsch Ges Inn Med 1986;92:503-8.
- Laxdal T, Jonasson TA. Adenine phosphoribosyltransferase deficiency in Iceland. Acta Med Scand 1988;224(6):621-6.
In our experience, 2,8-DHA crystals should be readily detected by routine urine microscopy in almost all patients with xanthine dehydrogenase. Occasionally, the crystals may be hard to identify, possibly due to decreased crystal clearance in some patients with severely reduced kidney function [1]. The small and medium sized crystals have a central Maltese cross pattern on polarized light microscopy whereas the large crystals do not as they are impermeable to light (see Figures 1a and 1b below).

More photos are available on the crystal photo reference page.
Diagnosis of nephrolithiasis requires radiologic imaging techniques that can detect radiolucent calculi such as ultrasound or computerized tomography. Analysis of 2,8-DHA crystals and stone material with infrared and ultraviolet spectrophotometry and/or x-ray crystallography easily differentiates 2,8-DHA from uric acid [2, 3]. In contrast, stone analysis employing biochemical methods, such as colorimetric reaction and thermogravimetric analysis, does not distinguish 2,8-DHA from uric acid and has resulted in the erroneous diagnosis of uric acid nephrolithiasis [1, 3].
Analysis of the residual APRT activity in red cell extracts is useful for the diagnosis of APRT deficiency but is not widely available in clical laboratories. The correct diagnosis from enzyme assays in red cell lysates can, however, be masked by a recent blood transfusion [4]. Genetic testing, identifying mutations in both copies of the APRT gene, confirms the diagnosis and is a practical approach in populations where the predominant mutations have been defined [1, 5]. Kidney biopsy is rarely necessary except in cases of unexplained renal insufficiency when 2,8-DHA crystals are not detected or identified in the urine (See Figure 2 below).
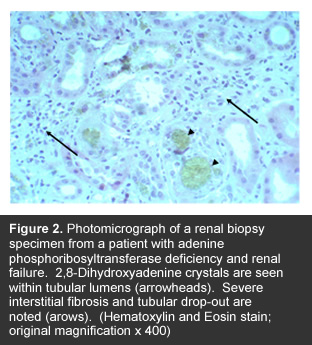 .
.
References
- Edvardsson V, Palsson R, Olafsson I, Hjaltadottir G, Laxdal T. Clinical features and genotype of adenine phosphoribosyltransferase deficiency in iceland. Am J Kidney Dis 2001;38(3):473-80.
- Laxdal T, Jonasson TA. Adenine phosphoribosyltransferase deficiency in Iceland. Acta Med Scand 1988;224(6):621-6.
- Gault MH, Simmonds HA, Snedden W, Dow D, Churchill DN, Penney H. Urolithiasis due to 2,8-dihydroxyadenine in an adult. N Engl J Med 1981;305(26):1570-2.
- Cassidy MJ, McCulloch T, Fairbanks LD, Simmonds HA. Diagnosis of adenine phosphoribosyltransferase deficiency as the underlying cause of renal failure in a renal transplant recipient. Nephrol Dial Transplant 2004;19(3):736-8.
- Suzuki K, Kobayashi S, Kawamura K, Kuhara T, Tsugawa R. Family study of 2,8-dihydroxyadenine stone formation: report of two cases of a compound heterozygote for adenine phosphoribosyltransferase deficiency (APRT*J/APRT*Q0). Int J Urol 1997;4(3):304-6.
The purine analog allopurinol, a potent inhibitor of xanthine dehydrogenase, has successfully been used for the treatment of APRT deficiency for the last 30 years [1, 2]. Allopurinol administered in the dose of 5-10 mg/kg/day (maximum suggested daily dose 600-800 mg), effectively prevents 2,8-DHA crystalluria and new stone formation and significantly improves kidney function in most patients with renal insuffiency [3, 4, 5]. Allopurinol appears to effectively prevent recurrent 2,8-DHA-induced nephropathy in the transplanted kidney [6-8]. Allopurinol is generally well tolerated although approximately 5% of patients experience adverse effects leading to the discontinuation of the drug [9]. In most cases these side effects are relatively mild and limited to gastrointestinal intolerance and skin rash. However, approximately 0.4% of patients develop allopurinol hypersensitivity syndrome which is characterized by fever, cutaneous manifestations, eosinophila and multiorgan involvement, including interstitial nephritis and hepatitis [2, 10]. Intolerance of allopurinol poses a serious problem for patients with APRT deficiency and, thus, alternative drug therapies are needed. Oxypurinol, an active metabolite of allopurinol, is also an inhibitor of xanthine dehydrogenase which has proven to be safe for the treatment of hyperuricemia and gout in patients allergic to allopurinol [9] but approximately 40% of patients who are allergic to allopurinol develop similar reactions to oxypurinol [9]. Additional xanthine dehydrogenase inhibitors are currently under study in animal models and humans [11]. Ample fluid intake, in the range of 2 to 2.5 L per day in adults and mild to moderate dietary purine restriction should be recommended to all patients with 2,8-dihydroxyadeninuria. Approximately 30% of patients with 2,8-DHA kidney stones require intervention for stone removal which is similar to patients with other types of kidney stone disease [3]. Extracorporeal shock-wave lithotripsy, lithotomy and endourological procedures are all effective for the treatment of acute stone episodes in patients with 2,8-DHA stones [3].
References
- Simmonds HA, Van Acker KJ, Cameron JS, Snedden W. The identification of 2,8-dihydroxyadenine, a new component of urinary stones. Biochem J 1976;157(2):485-7.
- Takano Y, Hase-Aoki K, Horiuchi H, et al. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci 2005;76(16):1835-47.
- Edvardsson V, Palsson R, Olafsson I, Hjaltadottir G, Laxdal T. Clinical features and genotype of adenine phosphoribosyltransferase deficiency in iceland. Am J Kidney Dis 2001;38(3):473-80.
- Gault MH, Simmonds HA, Snedden W, Dow D, Churchill DN, Penney H. Urolithiasis due to 2,8-dihydroxyadenine in an adult. N Engl J Med 1981;305(26):1570-2.
- Simmonds HA, Van Acker KJ, Cameron JS, Snedden W. The identification of 2,8-dihydroxyadenine, a new component of urinary stones. Biochem J 1976;157(2):485-7.
- Cassidy MJ, McCulloch T, Fairbanks LD, Simmonds HA. Diagnosis of adenine phosphoribosyltransferase deficiency as the underlying cause of renal failure in a renal transplant recipient. Nephrol Dial Transplant 2004;19(3):736-8.
- Brown HA. Recurrence of 2,8-dihydroxyadenine tubulointerstitial lesions in a kidney transplant recipient with a primary presentation of chronic renal failure. Nephrol Dial Transplant 1998;13(4):998-1000.
- Eller P, Rosenkranz AR, Mark W, Theurl I, Laufer J, Lhotta K. Four consecutive renal transplantations in a patient with adenine phosphoribosyltransferase deficiency. Clin Nephrol 2004;61(3):217-21.
- Wortmann RL. Recent advances in the management of gout and hyperuricemia. Curr Opin Rheumatol 2005;17(3):319-24.
- Gutierrez-Macias A, Lizarralde-Palacios E, Martinez-Odriozola P, Miguel-De la Villa F. Fatal allopurinol hypersensitivity syndrome after treatment of asymptomatic hyperuricaemia. Bmj 2005;331(7517):623-4.
- Yamada I, Fukunari A, Osajima T, Kamezawa M, Mori H, Iwane J. Pharmacokinetics/pharmacodynamics of Y-700, a novel xanthine oxidase inhibitor, in rats and man. Nucleosides Nucleotides Nucleic Acids 2004;23(8-9):1123-5.
Clinical Research
To date, no prospective studies have investigated the effect of allopurinol, or other treatment strategies, on the clinical outcome of patients with APRT deficiency. However, a retrospective study published by our group clearly shows that allopurinol prevents stone recurrence and even reverses established renal failure in properly treated patients.
Basic Research
Studies conducted in APRT knock-out mice revealed pathologic features similar to those found in human kidneys and the untreated animals developed recurrent nephrolithiasis and eventually died from renal failure [1]. In contrast, animals treated with allopurinol had no evidence of kidney damage [1-3]. These findings suggest that 2,8-DHA is directly nephrotoxic and plays a key role in the pathogenesis of kidney injury in affected patients.
References
- Redhead NJ, Selfridge J, Wu CL, Melton DW. Mice with adenine phosphoribosyltransferase deficiency develop fatal 2,8-dihydroxyadenine lithiasis. Hum Gene Ther 1996;7(13):1491-502.
- Engle SJ, Stockelman MG, Chen J, et al. Adenine phosphoribosyltransferase-deficient mice develop 2,8-dihydroxyadenine nephrolithiasis. Proc Natl Acad Sci U S A 1996;93(11):5307-12.
- Stockelman MG, Lorenz JN, Smith FN, et al. Chronic renal failure in a mouse model of human adenine phosphoribosyltransferase deficiency. Am J Physiol 1998;275(1 Pt 2):F154-63.
You can visit the APRT registry at the secure Mayo Clinic RedCAP site.
Our hypothesis is that APRT deficiency is significantly underdiagnosed in many countries, including in the US, and that the establishment of an international registry will facilitate the diagnosis and proper treatment of previously unidentified cases.
The goals of this project are:
- To collect clinical data into an international APRT deficiency registry in order to develop strategies for increasing the awareness and detection of the disease and, thus, improve clinical outcomes
- To determine the variables associated with stone recurrence and the efficacy of pharmacologic prevention
- To perform APRT mutation analysis in all participating patients who have not had an APRT mutation identified previously, study genotype-phenotype correlations and assess the role of urinary metabolic risk factors on the formation of kidney stones
- To interface with patient organizations, health care professionals and researchers through our APRT Deficiency Website to further enhance the educational mission of this project and disseminate knowledge to the community
- To develop a biobank for DNA and urine samples that can later be used for research, such as evaluation of the role of modifying genes.
We propose that this work will markedly reduce the suffering of affected patients and significantly reduce health care costs associated with APRT deficiency.
The team of investigators is well qualified to carry out this work, given the vast experience of individual team members in all aspects of APRT deficiency, including the diagnosis and management of the disorder, in addition to the large patient population that they have already generated and will be entered into the registry. Vidar Edvardsson, MD, is the Principal Investigator of the proposed study at Landspitali University Hospital (LUH) in Reykjavik. He is the Director of Pediatric Nephrology at the Children´s Medical Center and Coordinator of the Kidney Transplantation Program at LUH. Runolfur Palsson, MD, is a Co-Principal Investigator of the study. He is an Associate Professor of Medicine at the University of Iceland and Chief of the Division of Nephrology at LUH.
Patient Support
Currently, no patient support organizations devoted to APRT deficiency exist. However, if you or your physician or other health care providers have questions about the diagnosis of APRT deficiency, or care of patients who have this problem, please do not hesitate to contact Vidar Edvardsson, MD (vidare@landspitali.is) and/or Runolfur Palsson, MD (runolfur@landspitali.is) at Landspitali University Hospital in Reykjavik, Iceland (paging operator: 354 – 543-1000).
Photo Reference
High resolution photo micrographs of 2,8-DHA crystals are available as a reference for physicians and patients
More Information
More information on APRT deficiency can be located through the OMIM® - Online Mendelian Inheritance in Man® database.
 Consortium Home
Consortium Home APRT Deficiency
APRT Deficiency