
Dent Disease
Disease Information

Expand All Topics | Close All Topics
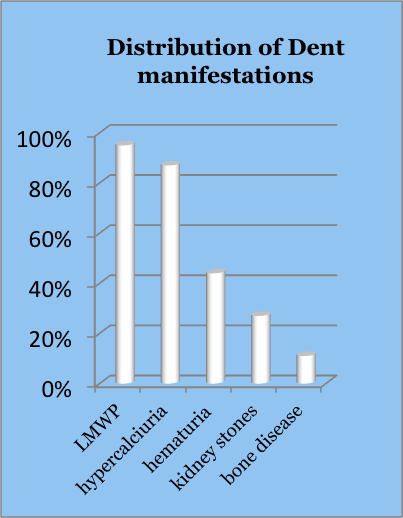
Dent disease (Dent's disease) is a rare X-linked disease that affects the kidneys of male patients. Typical manifestations include low molecular weight proteinuria, hypercalciuria, hematuria, nephrocalcinosis, kidney stones, osteomalacia, aminoaciduria and hypophosphatemia.
The clinical course varies greatly among patients, even within families with the same mutation.
Two genes are currently known to cause Dent disease, both located on the X-chromosome. Based on the gene involved, Dent disease can be divided into 2 types: Dent 1 and Dent 2.
Patients with Dent disease 1 have a mutation in CLCN5 and account for approximately 60% of cases. Patients with Dent disease 2 have a mutation in OCRL and account for approximately 15% of cases. Molecular genetic testing for both genes is currently available. Approximately 25% of patients do not appear to have mutations in CLCN5 or OCRL.
ORCL mutations can cause both Lowe Syndrome and Dent disease type 2. For reasons that are not currently understood, individuals with an OCRL mutation and Dent 2 phenotype lack key features of Lowe Syndrome such as dense cataracts and significant cognitive impairment.
- Foamy urine
- Increased thirst
- Pain or blood in the urine associated with the passage of kidney stones
- Signs and symptoms related to loss of kidney function including anemia, fatigue, loss of appetite and weight
- Signs and symptoms related to osteomalacia (weak bones) including boney pain.
Lab Findings
Common clinical features of Dent Disease (Dent's disease) include:
- Proteinuria in the range of 1-2 g per day, approximately half of which is low molecular weight (LMW; < 30,000 Da). Specific LMW proteins that can be tested include α1 microglobulin, retinol binding protein, and β2 microglobulin.
- Modest hypercalciuria, in the range of 4-6 mg/kg body weight is often observed as long as renal function is still good (GFR > 30 ml/min/1.73 m2)
- Low blood levels of parathyroid hormone (PTH)
- Increased blood levels of 1,25 vitamin D
- Normal levels of blood calcium
- Urinary oxalate and citrate levels are typically normal
- Nephrocalcinosis
- Increased urinary levels of amino acids
- Progressive renal failure (ESRD is observed in 30-80% of affected individuals by 30-50 y/o)
Other defects of proximal tubular function occurred in less than half of all patients, and include:
- Renal phosphate wasting and low levels of phosphate in the blood
- Renal potassium wasting
- High levels of glucose in the urine
- Kidney stones composed of calcium oxalate, calcium phosphate, or both together
- Rickets
- Short stature/growth delay
The overlap of many of these features with those seen in many patients afflicted with routine calcium oxalate kidney stones has often made it difficult to identify those patients affected with Dent Disease.
To facilitate diagnosis of Dent Disease, the Rare Kidney Stone Consortium in collaboration with leading Dent disease specialists has developed a diagnostic algorithm published in Pediatric Nephrology in 2013:
Edvardsson V, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani L. Milliner DS, and Palsson R. Hereditary Causes of Kidney Stones and Chronic Kidney Disease. Ped Neph 2013.
Article link: http://rd.springer.com/article/10.1007/s00467-012-2329-z
http://www.ncbi.nlm.nih.gov/pubmed/2333438
Genetic Screening
Patients can be definitively diagnosed if they have a mutation in one of the two genes known to cause this disorder, CLCN5 (Dent Disease 1) or OCRL1 (Dent 2).
This testing can be obtained through commercial laboratories (for example http://www.moldiag.de/en/gen/300008.htm; http://www.moldiag.de/en/gen/300535.htm), although insurance companies often will not reimburse for such disease-specific genetic testing.
Research Genetic Testing:
As part of our research protocol, the Rare Kidney Stone Consortium also performs genetic testing of patients and family members at risk of having the disease or being carriers.
For more information please contact rarekidneystones@mayo.edu.
Screening for Dent Disease and Dent Disease Carriers
The presence of low molecular weight proteins in the urine is usually detected by measuring levels of two common proteins: α1 microglobulin and retinol binding protein (RBP). Norden and colleagues demonstrated that the finding both an elevated level of urinary RBP and urine albumin consistently and reliably quantified and identified the low molecular weight proteinuria of affected male Dent patients [1]. Since Dent disease is X-linked, females can be carriers i.e., have one normal and one affected X chromosome). Although carrier females do not appear to any serious health consequences, they do often have urinary findings that are not quite normal. Since approximately one half of a carrier-female's sons will be affected, it is often desirable to screen for these subtle findings. Increased levels of urinary RBP appears to be the most sensitive marker when compared to β2 microglobulin, and α1 microglobulin, and successfully identified 21 of 24 (~88%) known carrier females (1).
See Gene Tests for complete discussion of the genetics of Dent disease
(http://www.ncbi.nlm.nih.gov/books/NBK99494/).
Reference
- Norden AG, Scheinman SJ, Deschodt-Lanckman MM, Lapsley M, et al. Tubular proteinuria defined by a study of Dent's (CLCN5 mutation) and other tubular diseases. Kidney Int 2000; 57: 240-249.
The mechanisms that cause kidney scarring and eventual kidney failure in Dent disease are poorly understood. Unfortunately, no published treatment trials are available to help physicians choose the best therapy. Available recommendations come from those doctors with the most experience caring for these patients. Since we assume that kidney calcification and kidney stones are caused by high levels of calcium in the urine, treatment typically includes a thiazide diuretic to increase renal calcium reabsorption and thereby reduce levels of calcium in the final urine. Although thiazide drugs can reduce levels of calcium in the urine of Dent disease patients, no information is available whether or not this can reduce the number of stones or protect kidney function. A published study in a mouse model of Dent disease suggested that a high citrate diet delayed loss of kidney function [1]. However there is no data on the use of citrate-containing medications in patients with Dent disease. For those Dent disease patients with osteomalacia (weak bones) vitamin D or related compounds have been used, and apparently this has improved bone strength in some [2]. However, use of vitamin D could also increase levels of calcium in the urine, and it has been suggested that the dose of Vitamin D be carefully titrated to the smallest amount needed to normalize serum levels of the bone hormone alkaline phosphatase [3]. Although not studied, patients are often prescribed ACE inhibitors or ARB's for reduction of proteinuria, with unknown effect on disease progression. Patients that develop renal failure have been given kidney transplants with good success [3].
References
- Cebotaru V, Kaul S, Devuyst O, Cai H, et al. High citrate diet delays progression of renal insufficiency in the ClC-5 knockout mouse model of Dent's disease. Kidney Int 2005; 68: 642-652.
- Wrong OM, Norden AGW, Feest TG. Dent's disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and marked male predominance. QJMed 1994; 87: 473-493.
- Scheinman SJ. X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int 1998; 53: 3-17.
Clinical Research
Phenotypic Heterogeneity and Identification of the Genetic Defect
After four groups independently described similar diseases with an X-linked pattern of inheritance, analysis of large affected kindreds were using a positional cloning approach and X-linked polymorphic genetic markers mapped the genetic defect to a region on the short arm of the X-chromosome (Xp11.22) in the North American patients [1] and the large Italian kindred [2]. A fortuitous microdeletion in a single affected child in Great Britain allowed the defect to be precisely mapped to ClCN5, which encodes a kidney-specific voltage gated chloride channel [3]. It was subsequently demonstrated that ClC-5 function as voltage-dependent chloride/proton exchanger [4, 5]. Disease causality for other patients was firmly established by documenting mutations (nonsense, missense, splice site and insertion/deletion) in other individuals with Dent disease and XRN [6]. Subsequently, patients in Italy (XRHR) and Japan (ILMWPHN) were all found to have mutations of the same gene [6, 7].
Therefore, all four diseases are now known to be phenotypic variants of mutations in the CLCN5 gene, and are collectively referred to as Dent Disease 1. Although proximal renal tubular dysfunction (Fanconi syndrome) is the predominant clinical phenotype in these four disorders, and hypercalciuria, nephrolithiasis, nephrocalcinosis and renal failure are unifying features, differences in disease expression between families and individuals remain poorly understood [8]. The same mutations were found to be associated with wide variations of clinical phenotypes, from classic Dent disease to isolated hypercalciuria, not only between unrelated patients but also within the same family [9]. Moreover, metabolic bone disease (rickets and osteomalacia) and renal failure are common in certain families but not others. The molecular basis for this observed phenotypic heterogeneity is not currently apparent. Low molecular weight proteinuria remains the most consistently observed abnormality.
In the original two cases of Dent and Friedman [10] the phenotype was characterized by hypercalciuria, rickets, tubular proteinuria, aminoaciduria, and phosphate wasting. In the series of Wrong and colleagues, end-stage renal disease developed in nine of fifteen affected males (~60%) and one of ten carrier females (~10%) at a mean age of 47 years [8]. Notably, one affected male did not require renal replacement therapy until his mid-sixties, and no other renal complications were documented in any other of the carrier female patients. Seven of eleven male patients (~64%) with nephrocalcinosis developed end-stage renal disease during a mean follow-up period of 24 years (range 6-34) but its presence was not a prerequisite for development of renal failure. Deterioration of renal function was still observed even in the absence of nephrocalcinosis [8]. Information regarding renal transplantation was available in three patients, two of whom received living-related allografts (father and brother) and the other a cadaveric renal allograft. Features suggestive of disease recurrence were present in the recipient of the cadaveric allograft and in the recipient of the paternal donor allograft [8]. In this series, low molecular weight proteinuria was found to be the most reliable disease marker, especially in carrier females, although absence of low molecular weight proteinuria cannot reliably exclude the disease since it was absent in some female carriers [8]. One-third of male and none of the female patients in this series developed bone disease [8].
There is evidence for genetic heterogeneity in Dent disease. In mutation analysis of 32 unrelated patients who met the clinical criteria for the diagnosis of Dent disease, 13 patients were found to have no mutation in the coding sequence of CLCN5. In one patient, the absence of mutation was confirmed by sequencing in two additional family members with clinical features of Dent disease [11]. It is unknown if these patients might have Dent 2 disease (see below) due to OCRL1 mutations, however recent studies suggest about 30% might [12, 13].
References
- Scheinman SJ, Pook MA, Wooding C, Pang JT, Frymoyer PA, Thakker RV: Mapping the gene causing X-linked recessive nephrolithiasis to Xp11.22 by linkage studies [see comments]. Journal.of.Clinical.Investigation. 91:2351-2357, 1993
- Bolino A, Devoto M, Enia G, Zoccali C, Weisserbach J, Romeo G: Genetic mapping in the Xp11.2 region of a new form of X-linked hypophosphatemic rickets. European Journal of Human Genetics 1:269-270, 1993
- Pook MA, Wrong O, Wooding C, Norden AGW, Feest TG, Thakker RV: Dent's disease, a renal Fanconi syndrome and nephrocalcinosis and kidney stones, is associated with a microdeletion involving DXS255 and maps to Xp11.22 by linkage studies. Human Molecular Genetics 2:2129-2134, 1993
- Picollo A, Pusch M: Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 436:420-423, 2005
- Scheel O, Zdebik AA, Lourdel S, Jentsch TJ: Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436:424-427, 2005
- Lloyd SE, Pearce SHS, Guenther W, Kawaguchi H, Igarashi T, Jentsch TJ, Thakker RV: Idiopathic low molecular weight proteinuria associated with hypercalciuric nephroclacinosis in Japanese children is due to mutations of teh renal chloride channel (CLCN5). J.Clin.Invest. 99:967-974, 1997
- Oudet C, Martin-Coignard D, Pannetier E, Champion G, Hanauer A: A second family with XLRH displays the mutation S244L in the CLCN5 gene. Hum.Genet. 99:781-784, 1997
- Wrong OM, Norden AGW, Feest TG: Dent's disease; a familial proximal renal tubualr syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and marked male predominance. Q.J.Med. 87:473-493, 1994
- Scheinman SJ, Cox JP, Lloyd SE, Pearce SH, Salenger PV, Hoopes RR, Bushinsky DA, Wrong O, Asplin JR, Langman CB, Norden AG, Thakker RV: Isolated hypercalciuria with mutation in CLCN5: relevance to idiopathic hypercalciuria. Kidney Int 57:232-239, 2000
- Dent CE, Friedman M: Hypercalciuric Rickets associated with renal tubular damage. Arch.Dis.Childh. 39:240-249, 1964
- Hoopes RR, Jr., Raja KM, Koich A, Hueber P, Reid R, Knohl SJ, Scheinman SJ: Evidence for genetic heterogeneity in Dent's disease. Kidney Int 65:1615-1620, 2004
- Hoopes RR, Jr., Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, Nussbaum RL, Scheinman SJ: Dent Disease with mutations in OCRL1. Am J Hum Genet 76:260-267, 2005
- Utsch B, Bokenkamp A, Benz MR, Besbas N, Dotsch J, Franke I, Frund S, Gok F, Hoppe B, Karle S, Kuwertz-Broking E, Laube G, Neb M, Nuutinen M, Ozaltin F, Rascher W, Ring T, Tasic V, van Wijk JA, Ludwig M: Novel OCRL1 mutations in patients with the phenotype of Dent disease. Am J Kidney Dis 48:942 e941-914, 2006
Basic Research
Dent Disease 1
The CLCN5 gene is a member of a large family of 10 genes encoding voltage-gated chloride channels that are structurally distinct from 2 other classes of chloride channels (the glycine and gamma-amino-butyric acid (GABAA) neuro-receptors and the cystic fibrosis transmembrane conductance regulator (CFTR)). Functional defects in the CLC-Ka and CLC-Kb chloride channels which belong to the same family as CLC-5 have been implicated in 2 other renal disorders, nephrogenic diabetes insipidus and Bartter's syndrome, respectively [1]. It has been recently found that CLC-5 is not a classic chloride channel but that it functions as electrogenic chloride/proton antiporter [2, 3]. The CLCN5 gene has 12 exons spanning approximately 30 kilobases of genomic DNA, and encodes a 746 amino acid protein (ClC-5) which has significant homology to all other identified members of the family of voltage-gated chloride channels [1]. The ClC-5 channel protein is kidney specific and expressed in different portions of the nephron, including the cortical proximal renal tubule, medullary thick ascending limb of Henle's loop and alfa-intercalated cells of the collecting duct [4]. To date, more than 80 separate mutations of the CLCN5 gene have been described consisting of nonsense, missense, splice site, insertional and deletional mutations [5]. When expressed in Xenopus oocytes, all mutations lead to either inactivation or severe reduction in chloride conductance [6]. More than 70% of mutations described to date are predicted to result in truncated or absent ClC-5, leading to complete loss of channel function [6]. Many of the remaining mutations affect the transmembrane domains and thereby abolish channel activity. Four mutations have been described that result in reduced Cl- channel conductance, and appear to cluster in a region of the protein that may have regulatory functions. However, there appears to be no correlation between phenotype and the type of mutation, or between phenotype and the degree of residual chloride conductance when mutant proteins are expressed in vitro. There are also some patients reported with clinical features suggestive of Dent disease without any documented mutations in the CLCN5 gene, supporting molecular heterogeneity. The mechanism(s) whereby the defective ClC-5 channel activity causes disease is incompletely understood [1]. Monospecific antibodies to ClC-5 localized the protein to early endosomes in proximal tubular cells, where it colocalized with endocytosed β2-microglobulin [7], and studies in a mouse transgenic mouse model suggest that disruption of the CLCN5 gene results in defective endocytosis and vesicle trafficking in proximal tubular cells [8]. It appears that the ClC-5 chloride/proton antiporter is critical for acidification of the endocytotic vesicles, which in turn is crucial for correct trafficking of membrane proteins [7]. CLC-5 knockout mice also exhibit loss of megalin and cubulin, multiligand, endocytic receptors that are colocalized in the renal proximal tubule and are heavily expressed in the apical endocytic apparatus [9]. It has also been found that C-terminal tail of ClC-5 interacts with cofilin, an actin-associated protein that is crucial in the regulation of albumin uptake by the proximal tubule [10]. In the mouse model, consequences of abnormal proximal tubular endocytosis include overexpression of the luminal PTH receptor [8]. It was postulated that consequent overstimulation of the PTH receptor could then result in excess hydroxylation of vitamin D and removal of the sodium-phosphate co-transporter NaPi-2 from the apical membrane, explaining the hypophosphatemia and hypercalciuria. The ClC-5 protein is also found in thick ascending limb cells [11], and it is possible that defective calcium reabsorption at this site could contribute to the hypercalciuria, although the potential mechanism is not understood.
Dent Disease 2
It has been recently found that some patients who had clinical Dent disease and no mutation of ClCN5 instead have mutations of the OCRL gene, which encodes a phosphatidylinositol 4,5-bisphosphate (PIP2) 5-phosphatase. This type of Dent disease has no major phenotypic difference from the previously described Dent disease, and it is now termed Dent 2 disease. Mutation of this gene located at Xq25 was initially found to cause Lowe oculocerebrorenal syndrome, which has some similar clinical findings to Dent disease. However, none of the reported mutations in Dent disease 2 were previously reported in patients with Lowe syndrome. Both diseases have significant LMW proteinuria, and can have other features of proximal tubular dysfunction such as glycosuria, aminoaciduria and phosphaturia. However, hypercalciuria, nephrocalcinosis and nephrolithiasis is common for Dent disease and very rare in Lowe syndrome. On the other hand, Lowe syndrome has prominent renal tubular acidosis, which commonly requires therapy, and which is not characteristic of Dent disease. Even more significant is the finding that Lowe syndrome always includes dense congenital cataracts, which are present to some extent even in carrier females; cataracts have never been described in Dent disease. Mild cognitive impairment was a feature in three of the five patients found to have this mutation that presented with a Dent phenotype, whereas it is present in a majority of patients with Lowe syndrome. These findings suggest a possibility of modifying loci and/or environmental factors that can alter phenotypic presentation of OCRL gene mutation [12]. Ludwig and colleagues recently published an additional series of patients with Dent 2 disease, and had found novel OCRL mutations in five of seventeen families with Dent disease phenotype who lacked a CLCN5 mutation. No systematic cognitive testing was done on this group of patients. Formal neuropsychological testing in patients with ClCN5-negative disease could be enlightening, although even patients with CLCN5 mutations have not been formally evaluated to date [13]. Both diseases are associated with decreased urinary megalin which can be found on the apical surface and in the endosomes of proximal tubular cells, implying that impaired apical membrane recycling could be a common defect underlying both conditions [14].
Dent Disease 1 vs. Dent Disease 2: Clues to Pathogenesis?
At this point, it is unclear how the mutations in Dent 1 (CLCN5) and Dent 2 (OCRL) produce a similar renal phenotype. However, there are some phenotypic differences that may shed some light. Elevated serum levels of muscle enzymes (LDH, CPK) is common in patients with OCRL mutations, but only seen in a few with CLCN5 mutations [13]. Conversely, although hypercalciuria is common to both, nephrocalcinosis is more common in patients with CLCN5 mutations [13]. Pooled data from larger series of patients will allow a more definitive determination of the relative phenotype, and allow hypotheses to be generated and then tested. For example, calcium and vitamin D were only rarely prescribed for Dents 2 patients, is this factor in their relative lack of nephrocalcinosis? Or do other urinary factors differ between the two groups (e.g., urinary supersaturation, citrate, glycoprotein inhibitor levels)
References
- Thakker RV: Pathogenesis of Dent's disease and related syndromes of X-linked nephrolithiasis. Kidney Int 57:787-793, 2000
- Picollo A, Pusch M: Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 436:420-423, 2005
- Scheel O, Zdebik AA, Lourdel S, Jentsch TJ: Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436:424-427, 2005
- Devuyst O, Christie PT, Courtoy PJ, Beauwens R, Thakker RV: Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a apthophysiological basis for Dent's disease. Hum.Moelc.Genet. 8:247-257, 1999
- Scheinman SJ, Cox JP, Lloyd SE, Pearce SH, Salenger PV, Hoopes RR, Bushinsky DA, Wrong O, Asplin JR, Langman CB, Norden AG, Thakker RV: Isolated hypercalciuria with mutation in CLCN5: relevance to idiopathic hypercalciuria. Kidney Int 57:232-239, 2000
- Yamamoto K, Cox JP, Friedrich T, Christie PT, Bald M, Houtman PN, Lapsley MJ, Patzer L, Tsimaratos M, Van THWG, Yamaoka K, Jentsch TJ, Thakker RV: Characterization of renal chloride channel (CLCN5) mutations in Dent's disease. J Am Soc Nephrol 11:1460-1468, 2000
- Gunther W, Luchow A, Cluzeaud F, Vandewalle A, Jentsch TJ: ClC-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc Natl Acad Sci U S A 95:8075-8080, 1998
- Piwon N, Gunther W, Schwake M, Bosl MR, Jentsch TJ: ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature 408:369-373, 2000
- Christensen EI, Devuyst O, Dom G, Nielsen R, Van der Smissen P, Verroust P, Leruth M, Guggino WB, Courtoy PJ: Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc Natl Acad Sci U S A 100:8472-8477, 2003
- Hryciw DH, Wang Y, Devuyst O, Pollock CA, Poronnik P, Guggino WB: Cofilin interacts with ClC-5 and regulates albumin uptake in proximal tubule cell lines. J Biol Chem 278:40169-40176, 2003
- Obermuller N, Gretz N, Kriz W, Reilly RF, Witzgall R: The swelling-activated chloride channel ClC-2, the chloride channel ClC-3, and ClC-5, a chloride channel mutated in kidney stone disease, are expressed in distinct subpopulations of renal epithelial cells. J Clin Invest 101:635-642, 1998
- Hoopes RR, Jr., Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, Nussbaum RL, Scheinman SJ: Dent Disease with mutations in OCRL1. Am J Hum Genet 76:260-267, 2005
- Utsch B, Bokenkamp A, Benz MR, Besbas N, Dotsch J, Franke I, Frund S, Gok F, Hoppe B, Karle S, Kuwertz-Broking E, Laube G, Neb M, Nuutinen M, Ozaltin F, Rascher W, Ring T, Tasic V, van Wijk JA, Ludwig M: Novel OCRL1 mutations in patients with the phenotype of Dent disease. Am J Kidney Dis 48:942 e941-914, 2006
- Ludwig M, Utsch B, Monnens LA: Recent advances in understanding the clinical and genetic heterogeneity of Dent's disease. Nephrol Dial Transplant 21:2708-2717, 2006
Due to its rarity, Dent disease expression has not been well defined, progress defining its pathophysiology has been slow, and there has been little opportunity to critically evaluate treatment interventions. Most nephrologists and urologists, the specialists most likely to be involved in the care of affected patients, have little or no exposure to these disorders during their training, and may participate in the care of only a few or no such patients during the course of a practicing lifetime. Limited clinical experience, and the restriction of definitive diagnostic methods to a small number of reference laboratories (e.g., detection of confirmed mutations for the causative genes CLCN5 and OCRL, or even measurement of low molecular weight urinary proteins), contribute to errors in diagnosis and suboptimal management of patients.
A centralized database can unify international research efforts and centralize information, allowing the tracking of larger numbers of patients than are currently accessible to individual researchers at single centers. The Dent registry will expand knowledge of the clinical expression of this disease by systematically accumulating and analyzing information regarding a larger number of patients than have been studied to date. The registry will define the spectrum of clinical expression, natural history of the disease, current treatments in use and the relative effectiveness. Data in the registry will allow development of consensus, evidence-based guidelines for diagnosis and management. and identify patient cohorts for clinical trials. This data can then be made readily available information for use by clinicians and biomedical researchers, who can use it to test new hypotheses. Does the degree of hypercalciuria correlate with renal outcome? Do thiazides appear to impact urine chemistries or patient outcome? Does the degree of low molecular weight proteinuria have prognostic significance? Are carrier females at risk for stones or CKD?
We are also developing a biobank of relevant samples including urine, blood, and DNA. A mutation database will be a long term goal of the registry that would promote structure-function analysis of CLCN5 or OCRL mutations and easy identification of sequence polymorphisms that have no phenotypic effect. This information will allow observations regarding the phenotype and prognosis of patients in each group. Differences might provide important clues regarding important pathogenic factors. Eventually, when sufficient data is obtained the effects of newly-discovered mutations can be predicted and modifier loci mapped. Detailed data on disease presentation, outcome, and other phenotypic features will be needed in order to use the mutation database for genotype-phenotype correlations. Data in the registry will be generally available to the biomedical community. A Dent Registry Scientific Review Panel will be responsible for review of all requests for data or biomaterials.
Publication list
A. Review papers
- Genetic causes of kidney stones and kidney failure. Lada Beara-Lasic , Vidar O. Edvardsson, Runolfur Palsson, John C. Lieske, David S. Goldfarb and Dawn S. Milliner. Clinical Reviews in Bone and Mineral Metabolism, January 2012.
- Hereditary causes of kidney stones and chronic kidney disease. Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, Palsson R. Pediatr Nephrol. 2013 Jan 20. [Epub ahead of print] PubMed PMID: 23334384.
B. Books and Chapters
- Lieske JC, Milliner DS, Beara-Lasic L, Rossetti S. Dent Disease. 2012 Aug 09. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews™ [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Available From http://www.ncbi.nlm.nih.gov/books/NBK99494/ PubMed PMID: 22876375.
C. Poster presentations at international meetings
- Presenting Characteristics of Dent Disease Patients. John C. Lieske, MD, FASN, Steven J. Scheinman, MD, FASN, Lawrence A. Copelovitch, MD, Hae Il Cheong, MD, PhD, Eric J. Bergstralh, Ramila A. Mehta and Lada Beara Lasic, MD.3rd Conference on Clinical Research for Rare Diseases, Oct 2012, Rockville, MD.
- Presenting Characteristics of Dent Disease Patients. John C. Lieske, MD, FASN, Steven J. Scheinman, MD, FASN, Lawrence A. Copelovitch, MD, Hae Il Cheong, MD, PhD, Eric J. Bergstralh, Ramila A. Mehta and Lada Beara Lasic, MD. Annual Meeting of The American Society of Nephrology, November 2012, San Diego, CA.
| Patients in Registry |
Age at diagnosis mean years |
Diagnosed after age 20 (%) | Type of mutation |
|---|---|---|---|
|
|
|
CLCN5 (Dent 1) 78 OCRL (Dent 2) 7 Non 1, Non 2 10 Unknown 10 |

Patient Advocacy Groups
A Dent Patient Advocacy Group (PAG) has been started and the new website is: http://dentdisease.com/
Further information regarding Lowe Syndrome may be found at the Lowe Syndrome Association website.
The following references may be helpful for more detailed information:
- Hoopes RR, Jr., Raja KM, Koich A, Hueber P, Reid R, Knohl SJ, Scheinman SJ: Evidence for genetic heterogeneity in Dent's disease. Kidney Int 65:1615-1620, 2004
- Utsch B, Bokenkamp A, Benz MR, Besbas N, Dotsch J, Franke I, Frund S, Gok F, Hoppe B, Karle S, Kuwertz-Broking E, Laube G, Neb M, Nuutinen M, Ozaltin F, Rascher W, Ring T, Tasic V, van Wijk JA, Ludwig M: Novel OCRL1 mutations in patients with the phenotype of Dent disease. Am J Kidney Dis 48:942 e941-914, 2006
- Hoopes RR, Jr., Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, Nussbaum RL, Scheinman SJ: Dent Disease with mutations in OCRL1. Am J Hum Genet 76:260-267, 2005
- Dent CE, Friedman M: Hypercalciuric Rickets associated with renal tubular damage. Arch.Dis.Childh. 39:240-249, 1964
- Wrong OM, Norden AGW, Feest TG: Dent's disease; a familial proximal renal tubualr syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and marked male predominance. Q.J.Med. 87:473-493, 1994
- Scheinman SJ: X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int. 53:3-17, 1998
- Ludwig M, Utsch B, Monnens LA: Recent advances in understanding the clinical and genetic heterogeneity of Dent's disease. Nephrol Dial Transplant 21:2708-2717, 2006
- Piwon N, Gunther W, Schwake M, Bosl MR, Jentsch TJ: ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature 408:369-373, 2000
- Christensen EI, Devuyst O, Dom G, Nielsen R, Van der Smissen P, Verroust P, Leruth M, Guggino WB, Courtoy PJ: Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc Natl Acad Sci U S A 100:8472-8477, 2003
- Hryciw DH, Wang Y, Devuyst O, Pollock CA, Poronnik P, Guggino WB: Cofilin interacts with ClC-5 and regulates albumin uptake in proximal tubule cell lines. J Biol Chem 278:40169-40176, 2003
- Norden AG, Scheinman SJ, Deschodt-Lanckman MM, Lapsley M, Nortier JL, Thakker RV, Unwin RJ, Wrong O: Tubular proteinuria defined by a study of Dent's (CLCN5 mutation) and other tubular diseases. Kidney Int. 57:240-249, 2000
- Cebotaru V, Kaul S, Devuyst O, Cai H, Racusen L, Guggino WB, Guggino SE: High citrate diet delays progression of renal insufficiency in the ClC-5 knockout mouse model of Dent's disease. Kidney Int 68:642-652, 2005
- Tosetto E, Ghiggeri GM, Emma F, Barbano G, Carrea A, Vezzoli G, Torregrossa R, Cara M, Ripanti G, Ammenti A, Peruzzi L, Murer L, Ratsch IM, Citron L, Gambaro G, D'Angelo A, Anglani F: Phenotypic and genetic heterogeneity in Dent's disease--the results of an Italian collaborative study. Nephrol Dial Transplant 21:2452-2463, 2006
Contact information:
Mailing address for records/paperwork:
Mayo Clinic
200 First Street SW
Rochester, MN 55905
Dent Disease Program Ei-SL 33
Email: rarekidneystones@mayo.edu
Phone: 800-270-4637
Fax: 507-255-0770
 Consortium Home
Consortium Home Dent Disease
Dent Disease