Expand All Topics | Close All Topics
Overview
Hyperoxaluria is a condition where too much of a substance called oxalate is present in the urine. Increased oxalate in the urine can come from eating too much oxalate in foods, from over-absorption of oxalate by the intestines due to certain diseases (called enteric hyperoxaluria), or to overproduction of oxalate in the body. In some persons the cause of the disease is not known, but may result from changes in the way kidneys handle normal amounts of body oxalate. Hyperoxaluria is uncommon, though can be found in up to 20 percent of individuals with kidney stones .In its many forms, it may be found among all ages, from infants to people in their 70s. The highest amounts of oxalate in the urine are seen in diseases in which the liver produces too much oxalate. This occurs in primary hyperoxaluria, which is a genetic disorder.
Oxalate is a substance that is naturally found in the human body. Most of the oxalate in the body is made by the liver. Oxalate can also enter the body from food that is eaten. There is no known need for oxalate by the human body. It is eliminated as waste primarily by the kidneys.
A person with hyperoxaluria has an overabundance of oxalate in the urine. The excess oxalate then binds with calcium in the urine to form kidney stones.

The main causes of hyperoxaluria are:
Dietary hyperoxaluria: Eating a diet of high oxalate content foods can cause high levels of oxalate in the urine. Generally, if diet changes are made the urine oxalate will then decrease.
Enteric hyperoxaluria: This can occur from the over-absorption of oxalate by the intestines, sometimes due to chronic diarrhea, intestinal disorders or intestinal surgeries.
Primary hyperoxaluria: (PH) is a rare genetic (inherited) disorder that is present at birth, and leads to an increase in oxalate production within the body. Unlike dietary or enteric hyperoxaluria, the amount of oxalate in the urine is not greatly affected by changes in dietary oxalate.
Primary hyperoxaluria
Three types of PH have been described and can be readily diagnosed. The common factor in all three types of PH is the increase in oxalate production in the liver. Each of the types of PH causes a decrease in activity of a specific enzyme in the liver. The decrease in the enzyme activity in each case changes how the liver makes oxalate, and a much larger amount of oxalate is produced.
The liver enzymes involved in the three types of PH are as follows:
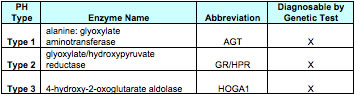
It is possible that other types of PH will be identified in the future. Some patients have high urine oxalates and kidney stones but do not have a genetic mutation that causes one of the three known types of PH. Future research may help us to better understand the cause of the high urine oxalate in these patients.
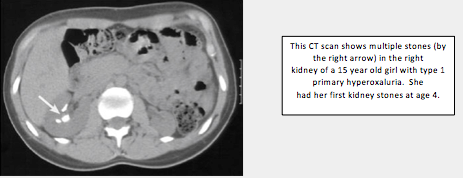
Primary hyperoxaluria (PH), types 1, 2, and 3, are rare genetic, or inherited, disorders that are present at birth. In a person with Type 1 primary hyperoxaluria, the liver creates too little of an enzyme called alanine-glyoxylate aminotransferase, or AGT. In Type 2, the liver is missing activity of a different enzyme, called glyoxylate reductase (GR) or hydroxypyruvate reductase. In Type 3 there is deficiency of hydroxy oxoglutarate aldolase. Very large amounts of oxalate are produced when there is not enough of these enzymes in the liver. Over time, oxalate in the urine can cause kidney stones. When very large amounts of oxalate are present in the urine, such as in primary hyperoxaluria, the kidneys can be damaged to the point that they quit working (kidney failure). Oxalosis happens after the kidneys fail and the excess oxalate builds up in the blood, and then spreads to the eyes, bones, muscles, blood vessels, heart and other major organs. If left untreated, PH can lead to serious illness and even death. Among patients with type 1 primary hyperoxaluria, about 50 percent will have kidney failure by age 30-35 years, Kidney failure happens less often in patients with types 2 but still occurs, and has not yet been described in patients with PH type 3. For that reason, it is critical that primary hyperoxaluria be diagnosed and treated as early as possible. Patients most often develop the first symptoms, typically kidney stones, anywhere from birth to the mid-20s. But hyperoxaluria may go unrecognized until age 30 to 40 years or even later. In some patients the first symptom is kidney failure.
PH is rare. It may be found among all ages, from infants to late adulthood. It is estimated that one to three people out of every million have PH.
Though high urine oxalate is present from birth, the symptoms of PH may first appear later. Symptoms most often begin in childhood or adolescence, though sometimes not until well into adulthood. Symptoms can range from mild to severe. The first sign of primary hyperoxaluria is typically symptoms related to kidney stones.
A 24 hour urine collection to determine the amount of urine oxalate is one of their first tests in evaluation of patients with kidney stones. Since there are other causes of high urine oxalate, however, the urine oxalate level alone will not confirm a PH diagnosis. A diagnosis of PH can now be confirmed in most patients through a blood test for genetic mutations (DNA) testing. Occasionally a patient might need a liver enzyme analysis done which would require a liver biopsy.
Patients will also have the details of their medical history, family history, diet and medication history evaluated. Additionally, patients should expect to undergo one or more of the following tests: Blood tests (this may include genetic (DNA) testing for both the patient and possibly family screening), urine testing , and radiology testing (x-ray, kidney ultrasound, or CT scan (computed tomography) in selected circumstances, liver or kidney biopsy, echocardiogram, or an eye examination may be recommended.
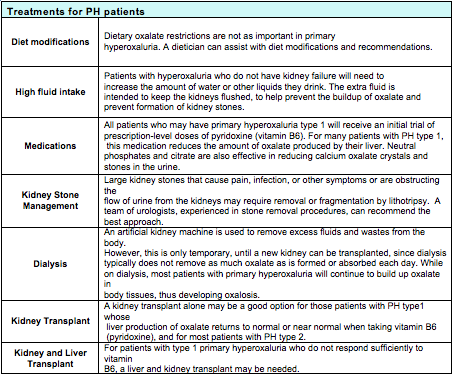
If PH is confirmed, treatments may include one or more of the following: high fluid intake, medications, diet modifications, kidney stone management. If there has been loss of kidney function, dialysis, kidney transplant, or liver transplant may be needed.
Signs and Symptoms
Hyperoxaluria may first appear any time from birth to adulthood, and symptoms can range from mild to severe. The first sign of hyperoxaluria is typically kidney stones. Patients that have a kidney stone get stuck in the urinary tract, are at risk of getting an infection.
Kidney stone symptoms may include:
- Abdominal pain – sudden or severe
- Pain when urinating
- Blood in their urine
- Urge to urinate often
- Fever and chills
Because kidney stones are uncommon in childhood, all children and adolescents who have symptoms of kidney stones or are diagnosed with kidney stones should be screened for hyperoxaluria. Pediatric patients with more severe hyperoxaluria can then be screened for primary hyperoxaluria via more specialized testing.
Primary hyperoxaluria that goes untreated may cause the kidneys to eventually be damaged and may stop working.
Signs of kidney failure include:
- A change in urine output – urine output is decreased or no urine is made
- Feeling generally ill, tiredness, and fatigued
- Nausea, vomiting
Sometimes, a patient may have already progressed to oxalosis and, during an eye examination, his or her ophthalmologist may discover oxalate crystals in the patient's eyes. Oxalosis in its late stages will cause bone disease, the result of oxalate crystals depositing in the bones and joints. It may also cause anemia that is difficult to treat, skin ulcers, and heart problems.
Diagnosis
Patients will hyperoxaluria or suspected of having PH will need a comprehensive physical exam, including a medical history, family history, and diet and medication history. Depending on their symptoms, patients may have one or more of the following tests to measure the presence and amount of oxalate in the body:
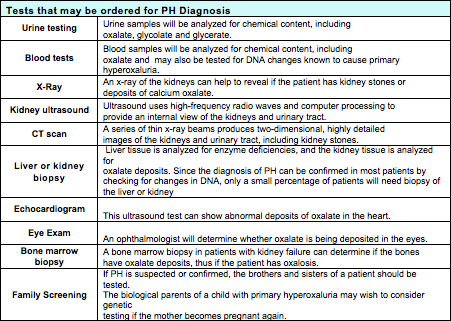
Tests such as blood levels of oxalate, urine or blood levels of glycolate or glycerate, liver biopsy enzyme analysis, and DNA testing are not generally available, but can be arranged through Mayo Medical Laboratories.
Treatments
Treatments for PH patients are individualized. Each patient is placed on a treatment program according to the type and severity of his/her disease.
The goals in treating PH patients are to keep the level of oxalate in the urine and the body as low as possible, to minimize the formation of kidney stones, and to maintain the patient's kidney health.
Treatment needs to be individualized for PH patients and needs to take into account each patient's needs, the type and severity of disease and how well the patient responds to treatment. Patients will need to be closely monitored over time by their medical team to measure their responses to the following treatments:
Research
Clinical Trials
PH Studies Open for Enrollment:
RKSC Primary Hyperoxaluria Registry
The Rare Kidney Stone Consortium is actively enrolling patients for the Primary Hyperoxaluria Registry. Following enrollment, information about the participant's medical history and findings will be collected and recorded by code number into the PH Registry. Updates to the information will be requested once each year thereafter. The collected data in this registry will help provide a better understanding of the how primary hyperoxaluria affects patients who have this condition. The goal of this registry is to collect data about this rare disease that may help lead the way to better treatments in the future.
While we have made good progress in the PH Registry enrollment, we are anxious to increase our enrollment and we invite all physicians to help! The larger the number of patients enrolled, the more we are able to learn about primary hyperoxaluria. Every patient counts! If you are interested in participating, please contact one of our Study Coordinators and they will make this possible.
Genetic testing for Primary Hyperoxaluria
This study involves a one-time blood draw that will be used to identify changes in DNA (mutations) that can cause primary hyperoxaluria. Disease expression, obtained from information in the PH Registry and medical records will be studied to see if there are certain patterns of disease that are associated with specific types of mutations. In addition, genetic causes of PH that is not related to mutations of the genes known to be involved in types 1, 2, and 3 PH – that is genetic causes of unclassified forms of PH – are being investigated. This study is open to patients with hyperoxaluria and their family members. The study will require a blood test.
Biobank
The purpose of this study is to make blood, urine and tissue samples available for use in research for the study of PH. Biobank samples will give scientists valuable research material that can help them to develop new diagnostic tests, treatments, and ways to prevent diseases.
Individuals who have been diagnosed with the PH will be asked to provide samples (blood, urine, or in some select cases tissue) that will be stored in a Biobank. The purpose of this study is to obtain high quality biologic samples from patients with PH and from their family members. These samples will be saved for use in future research.
The plan is to have 730 people take part in this multi-center study. The target goal of PH participants (patients or family members) is: 150 participants or family members who have primary hyperoxaluria.
Oxalate Absorption Study
The purpose of this study is to learn more about a condition called 'unclassified primary hyperoxaluria'. People with this condition have large amounts of oxalate in their urine, which can cause kidney stones and kidney failure. We do not know what causes the high level of oxalate in the urine in these patients. In this study, we will examine how oxalate taken with food affects this condition. Flavored gelatin (children) or a capsule (adults or children) containing 13C-labelled (a stable isotope) oxalic acid will be given to each subject along with a standard breakfast. Urine is collected for 24 hours following the breakfast and the amount of 13C oxalate is measured in the urine. We will compare the results of patients with unclassified primary hyperoxaluria to those of healthy volunteers.
For additional study information or to participate in the above studies:
Contact the study coordinator by phone or by e-mail.
Contact: Mayo Hyperoxaluria Center Study Coordinators: Julie Olson or Barb Seide
Phone: 800-270-4637
Email: hyperoxaluriacenter@mayo.edu
Laboratory Research
Laboratory research in primary hyperoxaluria is ongoing and includes multidisciplinary collaboration with physicians and researchers in Nephrology, Urology, Radiology, Laboratory Medicine and the basic sciences.
Patient Registry
RKSC Primary Hyperoxaluria Registry
The purpose of this registry is to identify as many individuals with primary hyperoxaluria as possible worldwide, and to collect as much clinical information about these patients as is possible. The resulting collection of data will be much larger than any individual center could hope to accumulate, and will be available to all interested physicians and researchers internationally in order to advance knowledge of this condition.
Goals of the registry are:
- To increase knowledge and understanding of primary hyperoxaluria
- To provide evidence that can be used to establish patient care guidelines
- To provide the basis for future clinical trials
Registry participation is voluntary and at the discretion of patients and their care providers. All information regarding individuals will be carefully protected in secure and confidential fashion in accordance with Mayo Clinic, United States, and international guidelines. This Registry's purpose is to identify as many individuals as possible around the world that are affected with PH, in order to determine how the disease behaves during the course of patients' lifetimes. It will be a great advantage for physicians and researchers to be able to study and compare the medical information on hundreds of PH patients instead of just a small number of patients known to a single medical center.
Because PH is a rare disease, even nephrologists and urologists who specialize in kidney diseases are likely to see only a very small number, often just 1 or 2, primary hyperoxaluria patients over the course of their entire career. That makes it very difficult to learn about how oxalate affects patient's kidneys and other body systems, and difficult to determine what treatments are effective. However, when doctors and patients share their experiences with PH and combine it with information from around the world, knowledge can advance much more rapidly.
The PH Registry is a computer database where such information is brought together, analyzed, used to advance understanding of the disease. Results are shared with physicians and scientists through publications and presentations, and with patients and family members through web publication of results.
Since the PH Registry has the ability to track a patient's health over a period of years and decades, the Registry's information will be valuable as physicians and researchers look for new methods to improve kidney function. The Registry will be useful in finding out which medications or treatments improve the health of PH patients.
The Registry has the ability to track a patient's health over a period of years and decades, the Registry's information will be valuable as physicians and researchers look for new methods to improve kidney function. The Registry will be useful in finding out which medications or treatments improve the health of PH patients.
RKSC Registry as of July 2015
What have we learned?
PH Type:There are at least 3 forms of primary hyperoxaluria.
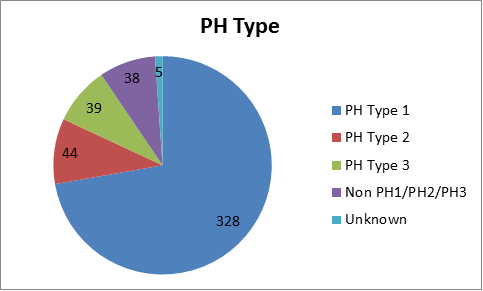
Of the 454 patients in the registry as of July 2015:
- 328 patients (72%) have primary hyperoxaluria Type 1
- 44 patients (10%) have primary hyperoxaluria Type 2
- 39 patients (9%) have primary hyperoxaluria Type 3
- 38 patients (8%) do not have known mutations for primary hyperoxaluria 1,2 or 3

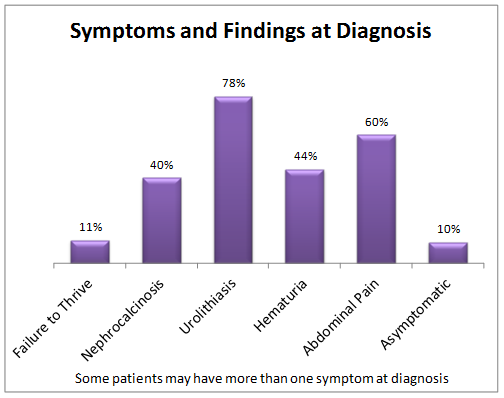
Of the 454 patients in the registry as of July 2015:
- 11% of the patients had failure to thrive (low height and weight) at diagnosis
- 40% of the patients had a history of nephrocalcinosis (when the kidneys are extensively filled with calcium oxalate crystals that can often cause loss of the kidneys.)
- 78% of the patients had a history of urolithiasis (kidney stones)
- 44% of the patients had a history of hematuria (blood in the urine)
- 60% of the patients had a history of abdominal or flank pain
- 10% of the patients were asymptomatic (had no symptoms at all) at diagnosis
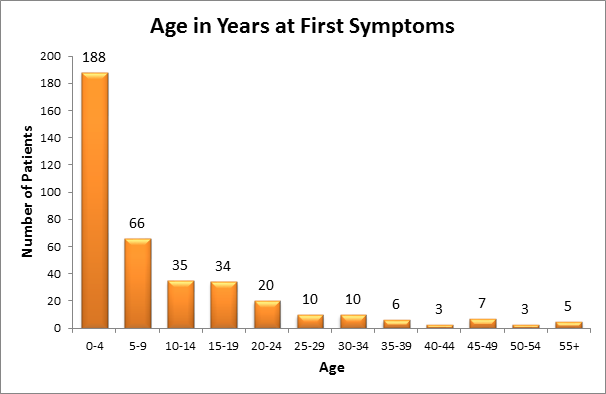 Of the 454 patients in the registry as of July 2015:
Of the 454 patients in the registry as of July 2015:
- Approximately 188 patients had their first symptom between the ages of 0-4 years
- Approximately 66 patients had their first symptom between the ages of 5-9 years
- Approximately 69 patients had their first symptom between the ages of 10-19 years
- Most patients have symptoms before the age of 25
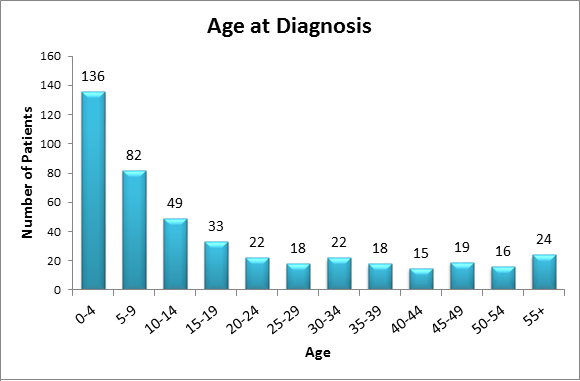 Of the 454 patients in the registry as of July 2015:
Of the 454 patients in the registry as of July 2015:
- 136 patients were diagnosed between the ages of 0-4 years
- 82 patients were diagnosed between the ages of 5-9 years
- The chart shows that a large percentage of diagnoses are made before a patient's 25th birthday
- Many patients are diagnosed before age 10; however other patients are not recognized to have PH until they are more than 40 years of age.
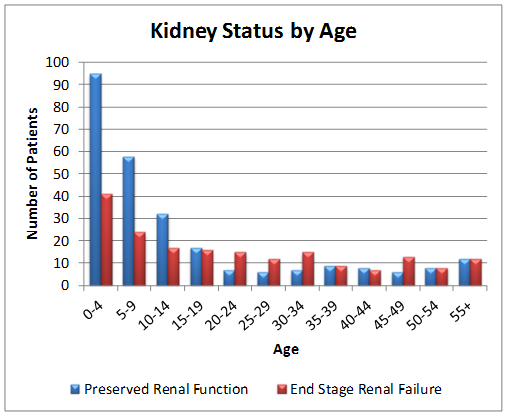 Of the 454 patients in the registry as of July 2015:
Of the 454 patients in the registry as of July 2015:
- The majority of patients have functioning kidneys at the time of diagnosis
Resources

Dawn S. Milliner, MD. (Principal Investigator) | bio
| Mayo Clinic, Rochester, MN



 Consortium Home
Consortium Home Primary Hyperoxaluria
Primary Hyperoxaluria